
News
Ein neuer Verdächtiger bei Chorea Huntington
23.08.2024 / Forschende aus Berlin und Düsseldorf haben mithilfe von Hirnorganoiden ein neues Gen mit dem Fortschreiten von Chorea Huntington in Verbindung gebracht. Das Gen trägt möglicherweise viel früher als bisher angenommen zu Anomalien im Gehirn bei, berichten sie in „Nature Communications“.
Erstmals haben Forschende das Gen CHCHD2 mit Chorea Huntington – einer unheilbaren, genetisch bedingten neurodegenerativen Erkrankung – in Verbindung gebracht und das Gen als mögliches therapeutisches Ziel identifiziert. In einem Hirnorganoid-Modell der Erkrankung haben sie festgestellt, dass Mutationen im Huntington-Gen HTT auch CHCHD2 beeinflussen; CHCHD2 spielt eine Rolle dabei, die normale Mitochondrienfunktion aufrecht zu erhalten. Die Studie wurde in „Nature Communications“ veröffentlicht.
An der Studie waren sechs verschiedene Labore des Max Delbrück Center unter der Leitung von Dr. Jakob Metzger von der Arbeitsgruppe „Quantitative Stammzell-Biologie“ und die Arbeitsgruppe „Stem Cell Metabolism“ von Professor Alessandro Prigione an der Medizinischen Fakultät der Heinrich-Heine-Universität Düsseldorf (HHU) beteiligt. Jedes Labor brachte sein einzigartiges Fachwissen zu Chorea Huntington, Hirnorganoiden, Stammzellenforschung und Genomeditierung ein.
„Es hat uns überrascht, dass Chorea Huntington die frühe Entwicklung des Gehirns durch Defekte, die mit mitochondrialer Fehlfunktionen zusammenhängen, beeinträchtigen kann“, sagt Dr. Pawel Lisowski, ein Erstautor aus Metzgers Arbeitsgruppe am Max Delbrück Center. „Das Organoidmodell deutet darauf hin, dass HTT-Mutationen die Gehirnentwicklung noch vor dem Auftreten klinischer Symptome schädigen. Es ist also sehr wichtig, diese spät auftretende neurodegenerative Erkrankung früh zu erkennen“, ergänzt Selene Lickfett, ebenfalls Erstautorin der Studie und Doktorandin in Prigiones Arbeitsgruppe an der HHU.
Drei Buchstaben werden ungewöhnlich oft wiederholt
Ein Organoid ist eine dreidimensionale, organähnliche Struktur, die Forschende im Labor aus Stammzellen entwickeln. Je nach Erkrankung und Fragestellung können Organoide aus unterschiedlichen Gewebearten gezüchtet werden. Sie sind nur wenige Millimeter klein, spiegeln jedoch das Zusammenspiel mehrerer Zelltypen wider. Kein anderes Modell aus der Petrischale liefert so komplexe Daten zu Zellfunktionen im menschlichen Körper.
Bei Chorea Huntington werden im Huntington-Gen HTT die drei Buchstaben für die Nukleotide Cytosin, Adenin, Guanin (CAG) ungewöhnlich oft wiederholt. Menschen mit 35 oder weniger Wiederholungen gehören im Allgemeinen nicht zur Risikogruppe, während 36 oder mehr Wiederholungen auf die Krankheit hindeuten. „Je größer die Anzahl an Wiederholungen, desto früher treten die Symptome der Krankheit auf“, erklärt Metzger, Letztautor der Studie. Die Mutationen lassen die Nervenzellen im Gehirn nach und nach absterben. Im Laufe der Zeit erleben Betroffene, dass sie ihre Muskeln immer weniger steuern können und sie entwickeln psychiatrische Symptome wie Impulsivität, Wahnvorstellungen und Halluzinationen. Weltweit sind etwa fünf bis zehn von 100.000 Menschen von Chorea Huntington betroffen. Aktuell verfügbare Therapien behandeln lediglich die Symptome; sie können weder den Verlauf verlangsamen noch die Krankheit heilen.
Das Gen HTT zu editieren, ist eine Herausforderung
Um die Auswirkungen von Mutationen im HTT-Gen auf die frühe Gehirnentwicklung zu studieren, nutzte Lisowski zuerst Varianten der CRISPR-Cas9-Geneditierungstechnik und manipulierte die DNA-Reparaturwege, um gesunde, induzierte pluripotente Stammzellen so zu modifizieren, dass sie eine große Anzahl an CAG-Wiederholungen enthielten. Dieser Prozess sei technisch herausfordernd, da Geneditierungswerkzeuge in Bereichen des Erbguts, die Sequenzwiederholungen wie die CAG-Wiederholungen in HTT enthalten, nicht effizient sind, sagt Lisowski.
Aus den genetisch veränderten Stammzellen züchteten die Forschenden dann Hirnorganoide – dreidimensionale Strukturen, die menschlichen Gehirnen in einem frühen Stadium der Entwicklung ähneln. Als sie die Genexpressionsprofile der Organoide in den verschiedenen Entwicklungsstadien analysierten, stellten sie fest, dass das CHCHD2-Gen durchgehend unterexprimiert war. Das wiederum reduzierte den Stoffwechsel der neuronalen Zellen. Bei gesunden Menschen gewährleistet CHCHD2 die Gesundheit der Mitochondrien – die Strukturen in den Zellen, die Energie produzieren. CHCHD2 wurde bisher mit Parkinson in Verbindung gebracht, aber noch nie mit Chorea Huntington.
Die Forschenden konnten die schädigende Wirkung auf die neuronalen Zellen rückgängig machen, wenn sie die Funktion des CHCHD2-Gens wiederherstellten. „Das war überraschend“, sagt Selene Lickfett. „Damit könnte dieses Gen prinzipiell ein Ziel für künftige Therapien sein.“
„Außerdem traten Defekte in neuronalen Vorläuferzellen und Hirnorganoiden auf, bevor sich potenziell toxische Aggregate des mutierten Huntingtin-Proteins gebildet hatten“, fügt Metzger hinzu. Dies deute darauf hin, dass die Pathologie im Gehirn bereits beginnt, lange bevor sie in der Klinik in Erscheinung tritt.
„Die vorherrschende Meinung ist, dass die Krankheit als Degeneration reifer Neuronen verläuft“, sagt Prigione. „Aber wenn sich die Veränderungen im Gehirn bereits früh im Leben entwickeln, müssen therapeutische Strategien möglicherweise zu viel früheren Zeitpunkten ansetzen.“
Weitreichende Implikationen
„Unsere Strategien zur Genomeditierung, vor allem, wenn sie die CAG-Wiederholungen im Huntington-Gen entfernen, sind sehr vielversprechend und konnten einige dieser Entwicklungsdefekte rückgängig machen. Das ebnet den Weg für eine potenzielle Gentherapie“, sagt Prigione. „Ein weiterer potenzieller Ansatz für die Therapien wäre es, die CHCHD2-Genexpression zu erhöhen.“
„Die Ergebnisse könnten auch breitere Anwendung bei anderen neurodegenerativen Krankheiten finden“, sagt Prigione. „Frühzeitige Behandlungen, die die hier gezeigten mitochondrialen Phänotypen rückgängig machen, könnten ein vielversprechender Ansatz für die Bekämpfung von altersbedingten Krankheiten wie Chorea Huntington sein.“
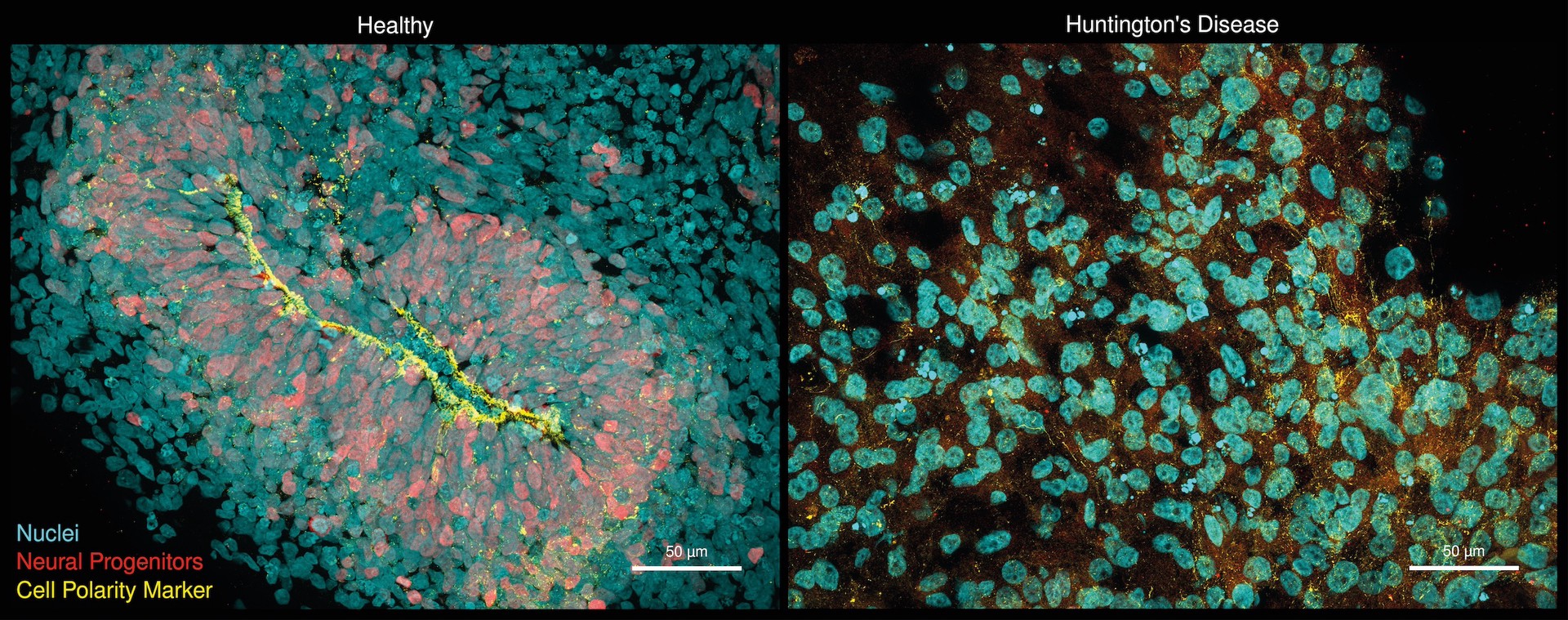
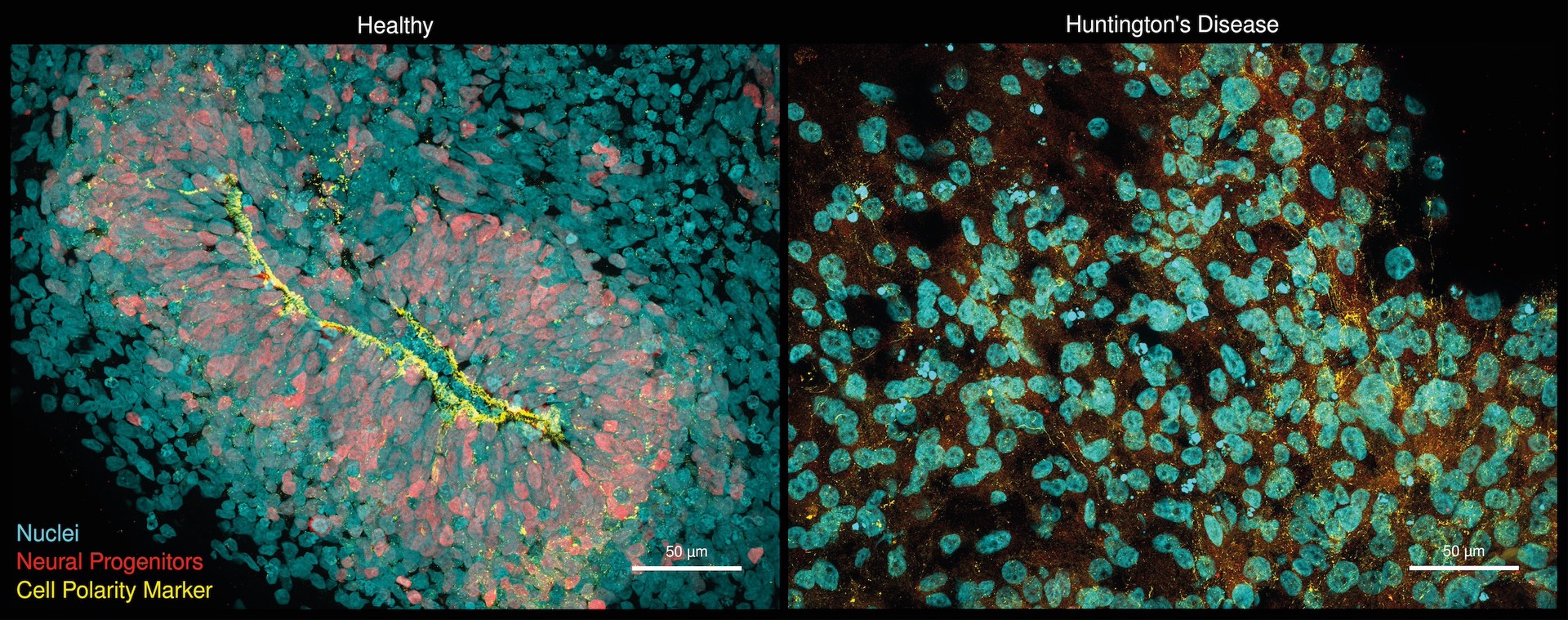
Hirn-Organoide, die aus induzierten pluripotenten Stammzellen entwickelt wurden: Gesunde Organoide (links) zeigen eine organisierte neurogene Zone (gelb) und neuronale Vorläuferzellen (rot). Organoide, die von der Huntington-Krankheit betroffen sind (rechts), weisen dagegen fast keine neurale Vorläuferzellen und zelluläre Polaritätsdefekte (gelb) auf. Diese Defekte sind in der Literatur ähnlich für menschlichen Föten mit der Huntington-Krankheit beschrieben. Foto: Selene Lickfett, Heinrich-Heine-Universität Düsseldorf, Werner Dykstra, Max Delbrück Center
{kind=link}
Das Max-Delbrück-Centrum für Molekulare Medizin in der Helmholtz-Gemeinschaft (Max Delbrück Center) gehört zu den international führenden biomedizinischen Forschungszentren. Nobelpreisträger Max Delbrück, geboren in Berlin, war ein Begründer der Molekularbiologie. An den Standorten in Berlin-Buch und Mitte analysieren Forscher*innen aus rund 70 Ländern das System Mensch – die Grundlagen des Lebens von seinen kleinsten Bausteinen bis zu organ-übergreifenden Mechanismen. Wenn man versteht, was das dynamische Gleichgewicht in der Zelle, einem Organ oder im ganzen Körper steuert oder stört, kann man Krankheiten vorbeugen, sie früh diagnostizieren und mit passgenauen Therapien stoppen. Die Erkenntnisse der Grundlagenforschung sollen rasch Patient*innen zugutekommen. Das Max Delbrück Center fördert daher Ausgründungen und kooperiert in Netzwerken. Besonders eng sind die Partnerschaften mit der Charité – Universitätsmedizin Berlin im gemeinsamen Experimental and Clinical Research Center (ECRC) und dem Berlin Institute of Health (BIH) in der Charité sowie dem Deutschen Zentrum für Herz-Kreislauf-Forschung (DZHK). Am Max Delbrück Center arbeiten 1800 Menschen. Finanziert wird das 1992 gegründete Max Delbrück Center zu 90 Prozent vom Bund und zu 10 Prozent vom Land Berlin. www.mdc-berlin.de
Hirn-Organoide, die aus induzierten pluripotenten Stammzellen entwickelt wurden: Gesunde Organoide (links) zeigen eine organisierte neurogene Zone (gelb) und neuronale Vorläuferzellen (rot). Organoide, die von der Huntington-Krankheit betroffen sind (rechts), weisen dagegen fast keine neurale Vorläuferzellen und zelluläre Polaritätsdefekte (gelb) auf. Diese Defekte sind in der Literatur ähnlich für menschlichen Föten mit der Huntington-Krankheit beschrieben. Foto: Selene Lickfett, Heinrich-Heine-Universität Düsseldorf, Werner Dykstra, Max Delbrück Center
Quelle: Gemeinsame Pressemitteilung der Heinrich-Heine-Universität Düsseldorf und des Max Delbrück CenterEin neuer Verdächtiger bei Chorea Huntington