
News
Mit Organoiden erstmals das Leigh-Syndrom erklären
22.04.2021 / Wenn die Kraftwerke der Zellen nicht richtig funktionieren, ist die Lebenswartung gering. Sonst weiß man wenig über das Leigh-Syndrom. Mit Organoiden haben Forschende aus Düsseldorf und Berlin nun ein erstes menschliches Modell dieser seltenen Erkrankung geschaffen und stellen es in „Nature Communications“ vor.
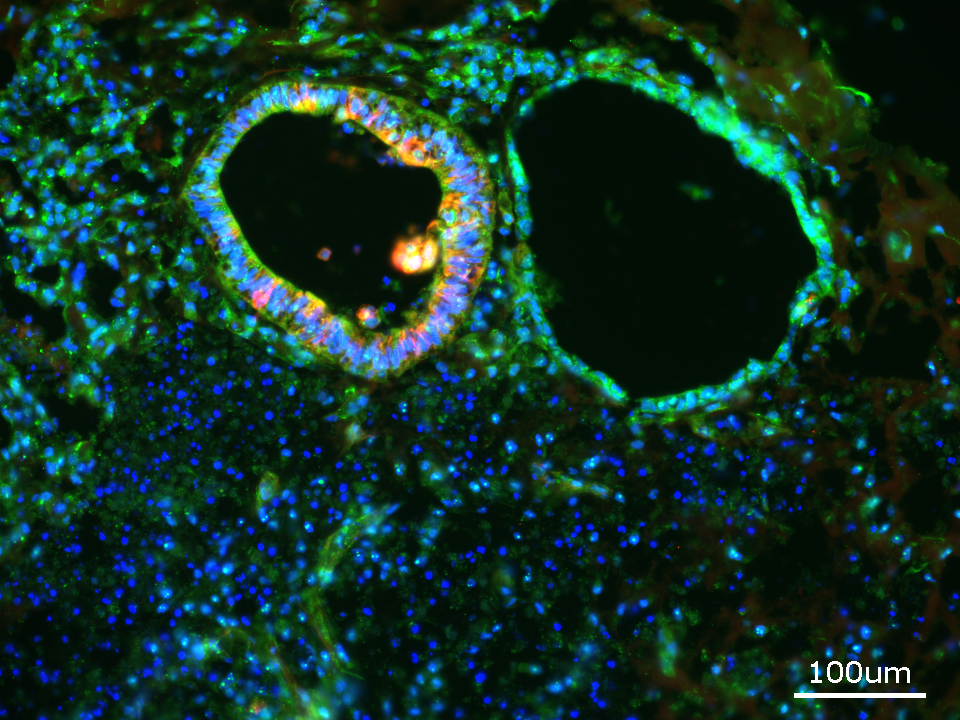
Das Leigh-Syndrom, auch als subakute nekrotisierende Enzephalomyelopathie bezeichnet, ist eine der schwersten erblichen Hirnerkrankungen bei Kindern. Dabei bringen verschiedene Genmutationen die Mitochondrien aus dem Gleichgewicht, die eine wichtige Rolle im Energiehaushalt des Körpers spielen. SURF1 ist eines der Kernene, das beim Leigh-Syndrom häufig verändert ist. Die betroffenen Kinder erkranken meist in den ersten Lebensmonaten, leiden an Bewegungsstörungen, Atem- und Schluckbeschwerden sowie einergeistigen Behinderung. Die meisten Kinder sterben innerhalb weniger Monate oder Jahre. Therapien gibt es nicht.
Gesucht: Angriffspunkte für gezielte Therapie
„Bislang gab es kein effektives Modell für das durch SURF1-Defekte verursachte Leigh-Syndrom, das Wissenschaftler*innen dabei unterstützt, die molekularen Mechanismen der Krankheit zu verstehen“, erklärt Professor Alessandro Prigione, der mit seinem Team an der Klinik für Allgemeine Pädiatrie des Universitätsklinikums Düsseldorf den Stoffwechsel von Stammzellen erforscht. Gemeinsam mit den Gruppen von Professor Nikolaus Rajewsky, Direktor des Berliner Instituts für Medizinische Systembiologie (BIMSB) des Max-Delbrück-Centrums für Molekulare Medizin in der Helmholtz-Gemeinschaft (MDC), und von Professor Markus Schuelke an der Klinik für Neuropädiatrie der Charité – Universitätsmedizin Berlin haben die Wissenschaftler*innen um Prigione nun das erste Organoid des Leigh-Syndroms entwickelt. Das Modell, das sie im Fachblatt „Nature Communications“ vorstellen, basiert auf induzierten pluripotenten Stammzellen (iPS-Zellen), die mit der CRISPR-Cas9-Genschere verändert wurden. Im Fachblatt „Nature Communications“ stellen sie es vor. „Wir zeigen mit dieser Studie, dass Modellsysteme aus iPS-Zellen von Patient*innen den Weg zu Therapien für eine seltene Krankheit mit hohem medizinischen Bedarf eröffnen“, umschreibt Erstautorin Dr. Gizem Inak die Bedeutung ihrer Arbeit.
Wissenschaftler*innen haben in der Vergangenheit vielfach versucht, mitochondriale Erkrankungen an Tiermodellen zu untersuchen. „Doch das Erbgut der Mitochondrien unterscheidet sich deutlich von der Kern-DNA“, erklärt Prigione. „Methoden, mit denen Gene im Zellkern modifiziert werden können, funktionieren bei Mitochondrien einfach nicht.“ Schon vor einigen Jahren hat Prigione, damals Delbrück-Fellow am MDC, mit seinem Forschungsteam ein zellbasiertes Modell für mitochondriale Erkrankungen entwickelt. Die Wissenschaftler*innen versetzten Hautzellen von Erkrankten in einen ursprünglicheren Zustand zurück und schufen so iPS-Zellen. Diese wandelten sie in einem weiteren Schritt in Nervenzellen um.
Neue Einsatzgebiete für vorhandene Medikamente finden
Dabei stellten sie fest, dass die Vorläuferzellen von Neuronen auf den Stoffwechsel der Mitochondrien angewiesen sind. Da dieser Stoffwechsel medikamentös beeinflusst werden kann, eignen sich die Vorläuferzellen als Modell für die Entwicklung von Medikamenten. So kann an den Vorläuferzellen getestet werden, wie verschiedene Substanzen wirken. Als Koordinator eines Konsortiums, das vom European Joint Program for Rare Diseases (EJP-RD) finanziert wird, sucht Prigione mit dieser Methode nach Wirkstoffen, die für andere Indikationen bereits zugelassen sind, möglicherweise aber auch gegen das Leigh-Syndrom wirken. Der Vorteil: Bei Medikamenten, die von den Zulassungsbehörden bereits geprüft worden sind, ist die Testphase deutlich kürzer. Klinische Studien können eher beginnen, so dass neue Therapien schneller zur Verfügung stehen.
Mit Organoiden die Hirnentwicklung modellieren
Auch für die aktuelle Studie schuf Gizem Inak aus den Zellen von Patient*innen iPS-Zellen. Mithilfe der Genschere CRISPR-Cas9 korrigierte sie die SURF1-Mutation. Parallel dazu schleuste sie das mutierte SURF1-Gen in gesunde Kontroll-iPS-Zellen ein. Dann gingen die Wissenschaftler*innen einen Schritt weiter. Dr. Agnieszka Rybak-Wolf, die die MDC-Technologieplattform „Organoide“ leitet, züchtete aus den genetisch veränderten Stammzellen Organoide. „Organoide ermöglichen es, die komplexe menschliche Gehirnentwicklung bis zu einem gewissen Grad zu modellieren. Mit Standard-Monolayer-Zellkulturen allein ist das nicht wirklich möglich“, sagt Agnieszka Rybak-Wolf. Organoide sind Miniatur-Organe, kaum so groß wie ein Stecknadelkopf, in denen sich die Zellen dreidimensional anordnen und einige Strukturen des Original-Organs in der Petrischale widerspiegeln – in diesem Fall der Gehirne von Menschen mit und ohne Leigh-Syndrom.
So entdeckten die Wissenschaftler*innen, dass die neuronalen Defekte beim Leigh-Syndrom möglicherweise von einem Energiedefizit bereits auf der Ebene der Vorläuferzellen verursacht werden. „Die Vorläuferzellen der Patient*innen bildeten keine Verzweigungen und differenzierten nicht weiter zu Neuronen“, beschreibt Prigione die Stagnation auf Ebene der Neuronen-Vorstufe. „Es war unglaublich, was in der Petrischale passiert ist, nachdem die Mutation korrigiert worden war“,sagt Agnieszka Rybak-Wolf. „Das Organoid fing an, sich zu entwickeln. Am Ende sah es fast genauso aus wie die gesunden Organoide.“
Leigh-Syndrom ist eine neurologische Entwicklungsstörung
Das war nicht die einzige Überraschung. Das Leigh-Syndrom galt bislang als neurodegenerative Erkrankung, ausgelöst von einer Schädigung der Neuronen durch freie Radikale. „Indem wir nachgewiesen haben, dass der gestörte Zellstoffwechsel bereits die neuronalen Vorläuferzellen beeinträchtigt, haben wir gezeigt, dass es sich beim Leigh-Syndrom nicht ausschließlich um eine neurodegenerative Erkrankung handelt, bei der sich Neuronen erst ausbilden und dann absterben“, fasst Gizem Inak zusammen. „Es handelt sich eher um eine neurologische Entwicklungsstörung.“ Dies könnte auch erklären, warum Kinder, die am Leigh-Syndrom erkrankt sind, häufig einen vergleichsweise kleinen Schädel haben oder geistig beeinträchtigt sind.
Ansätze für neue Behandlungsstrategien
„Dieser Einblick in den Mechanismus der Krankheit kann nun dafür genutzt werden, zielgerichtete Behandlungsstrategien für Kinder zu entwickeln, die an dieser seltenen Erkrankung leiden“, sagt Prigione. Ein Weg könnte darin bestehen, die verminderte Energieleistung in den Vorläuferzellen mithilfe der SURF1-Gentherapie zu verbessern. Dabei würde nicht das mutierte SURF1-Gen entfernt, sondern eine gesunde Kopie des Gen eingeschleust, damit sie die Mutation schachmatt setzt. Für andere neurologische Erkrankungen wird dieser Ansatz bereits getestet. Als möglichen Kandidaten für eine medikamentöse Therapie identifizierten die Wissenschaftler*innen Bezafibrat, ein Medikament, das zur Diätunterstützung bei Erwachsenen zugelassen ist. Es kurbelt nicht nur den Fettstoffwechsel an, sondern aktiviert auch das sogenannte PGC1-Alpha, einen Regulator des Zellstoffwechsels. „Ein Krankheitsmodell zu entwickeln war der erste Schritt“, fasst Prigione zusammen, „jetzt können wir dieses Modell nutzen, um nach den effektivsten therapeutischen Strategien gegen diese schwere Kinderkrankheit zu suchen.“
Weitere Informationen
- Dr. Agnieszka Rybak-Wolf, Technologieplattform Organoide
- AG Prigione, Stammzell Metabolismus
- Personalisierte Wirkstoffsuche ermöglicht Bekämpfung seltener Krankheiten
- Alessandro Prigione: Ein Modell seltener Krankheiten
Literatur
Gizem Inak et al: Defective metabolic programming impairs early neuronal morphogenesis in neural cultures and an organoid model of Leigh Syndrome”, in Nature Communications, DOI: 10.1038/s41467-021-22117-z
Das Max-Delbrück-Centrum für Molekulare Medizin in der Helmholtz-Gemeinschaft (MDC) wurde 1992 in Berlin gegründet. Es ist nach dem deutsch-amerikanischen Physiker Max Delbrück benannt, dem 1969 der Nobelpreis für Physiologie und Medizin verliehen wurde. Aufgabe des MDC ist die Erforschung molekularer Mechanismen, um die Ursachen von Krankheiten zu verstehen und sie besser zu diagnostizieren, verhüten und wirksam bekämpfen zu können. Dabei kooperiert das MDC mit der Charité – Universitätsmedizin Berlin und dem Berlin Institute of Health (BIH) sowie mit nationalen Partnern, z.B. dem Deutschen Zentrum für Herz-Kreislauf-Forschung (DHZK), und zahlreichen internationalen Forschungseinrichtungen. Am MDC arbeiten mehr als 1.600 Beschäftigte und Gäste aus nahezu 60 Ländern; davon sind fast 1.300 in der Wissenschaft tätig. Es wird zu 90 Prozent vom Bundesministerium für Bildung und Forschung und zu 10 Prozent vom Land Berlin finanziert und ist Mitglied in der Helmholtz-Gemeinschaft deutscher Forschungszentren. www.mdc-berlin.de
Quelle: Max Delbrück Center for Molecular Medicine in the Helmholtz Association (MDC)
Max-Delbrück-Centrum für Molekulare Medizin (MDC)